Distributors
BD manufactures and sells a wide selection of products and services for the healthcare, consumer, life sciences and industrial markets. Most of our products are sold through distributors and wholesalers.

- Distributor List
- Unique Device Identifier (UDI)
Distributor Names
 Browse All Distributors
Browse All Distributors
- Check your spelling
- Try different filter criteria
- Fine-tune your search with more keywords
- If an exact phase doesn’t work, try to search for similar words and pages
UDI compliance
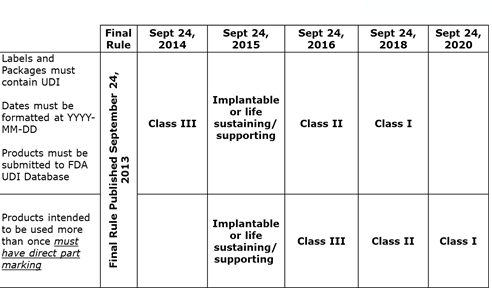
On September 24, 2013, the U.S. Food and Drug Administration (FDA) released a final rule requiring that most medical devices distributed in the United States carry a unique device identifier (UDI). The UDI system facilitates medical device identification, traceability and tracking through distribution and use.
The UDI system consists of three core segments. The first is the UDI, which is a unique number assigned to the version or model of a device that is required on all packaging labeling of finished goods. The UDI must include a device identifier (DI) and may contain a production identifier (PI). UDIs appear on labels in both plain text format and a format readable by automatic identification data capture (AIDC) technology (e.g., a barcode). This identifier also includes production-specific information such as the product's lot or batch number, expiration date and manufacturing date when that information appears on the label. Additionally, the date format must read as "YYYY-MM-DD" on all human-readable text (which does not include barcode text).
The second component is Direct Part Marking, which is a permanent UDI marking on the device. If a device will be used more than once and be reprocessed before each use, it must also be directly marked with a UDI, allowing accurate identification even when the device is no longer accompanied by its label or package.
The third component is the Global Unique Device Identification Database (GUDID), which is a publicly searchable database administered by the FDA that serves as a reference catalog for every device with an identifier.
UDI compliance timeline
The FDA defined a phased approach to compliance depending on the device's risk classification (reference the FDA UDI timeline chart).
BD requires that all of our suppliers implement and adhere to the FDA UDI timeline, promptly responding to queries and regular communication on UDI efforts.
Visit Unique Device Identifier (UDI) or AccessGUDID on the FDA website, or contact your BD representative for more information on the UDI system.
In 2013, the U.S. Food and Drug Administration (FDA) released a final rule establishing a unique device identification system designed to adequately identify devices through distribution and use. The final rule requires device labelers to include a unique device identifier (UDI) on device labels and packages, except where the rule provides for an exception or alternative. Each UDI must be provided in a plain text version and in a form that uses automatic identification and data capture (AIDC). technology. The UDI will also need to be directly marked on a device intended for more than one use, and intended to be reprocessed before each use. Dates on device labels and packages are to be presented in a standard format consistent with international standards and international practice.
A UDI is a unique numeric or alphanumeric code that consists of the following two parts:
1.A device identifier (DI), a mandatory, fixed portion of a UDI that identifies the labeler and the specific version or model of a device
2.A production identifier (PI), a conditional, variable portion of a UDI that identifies one or more of the following details when included on the label of a device:
- The lot or batch number a device was manufactured within
- The serial number of a specific device
- The expiration date of a specific device
- The date a specific device was manufactured on
- The distinct identification code required by §1271.290(c) for a human cell, tissue, or cellular- and tissue-based product (HCT/P) regulated as a device1
This final rule will substantially reduce existing obstacles to the adequate identification of medical devices used in the United States. By making it possible to rapidly and definitively identify a device and key attributes that affect its safe and effective use, the rule will reduce medical errors that result from the misidentification of a device or confusion concerning its appropriate use. The identification system established under this rule will lead to more accurate reporting of adverse events by making it easier to identify the device prior to submitting a report. It will allow the FDA, healthcare providers and the industry to more rapidly extract useful information from adverse event reports, pinpoint the particular device at issue and thereby, gain a better understanding of the underlying problems, and take appropriate, better-focused, corrective action. The rule will also require dates on medical device labels to conform to a standard format to ensure those dates are unambiguous and clearly understood by device users.
In 2013, the FDA released a final rule establishing a unique device identification system designed to adequately identify devices through distribution and use. The final rule requires device labelers to include a UDI on device labels and packages, except where the rule provides for an exception or alternative. Each UDI must be provided in a plain text version and in a form that uses automatic identification and data capture (AIDC) technology. The UDI is also required to be directly marked on a device intended for more than one use and intended to be reprocessed before each use.
No, BD will continue to use catalog numbers in addition to GS1 Global Trade Item Numbers (GTINs) for device identification.
Yes. BD ordering processes will continue to utilize the current BD catalog numbers―the ordering process will remain unchanged if you prefer to order by BD catalog numbers.
If you are interested in using the DI (GTIN) for your electronic data interchange (EDI) ordering processes, contact your BD representative.
Yes. In addition to using the BD catalog number, BD has started enhancing invoices or packing slips to include both DI and PI information.
Contact your BD representative for specific information or visit AccessGUDID.
BD produces all classes of medical devices. We also manufacture products that are exempt from the FDA UDI regulation, such as pharmaceutical products and research use only (RUO) products.
Yes. BD has already started the process of populating the FDA Global Unique Device Identification Database (GUDID), which is a repository for product information. BD Class III products are already in the GUDID. Additional products will be added to the GUDID on or before the FDA UDI timeline.
Generally, you will find that new BD labels will be enhanced with additional data and will continue to function in your scanning processes. In the event that a DI or PI (and the associated catalog number and barcode) change as result of BD UDI implementation, you will be notified.